Commands
Launch
The launch command opens one or more datasets in h5ad, loom, Seurat, TileDB, zarr, or STAR-Fusion formats. Results are stored on disk in JSON format.
usage: cirro launch [-h] [--spatial [SPATIAL ...]] [--markers [MARKERS ...]]
[--host HOST] [--port PORT] [--no-open]
[--results RESULTS] [--ontology ONTOLOGY]
[--tmap [TMAP ...]]
dataset [dataset ...]
Positional Arguments
- dataset
Path(s) to dataset in h5ad, loom, Seurat, TileDB, zarr, or STAR-Fusion format. Separate multiple datasets with a comma instead of a space in order to join datasets by cell id
Named Arguments
- --spatial
Directory containing 10x visium spatial data (tissue_hires_image.png, scalefactors_json.json, and tissue_positions_list.csv) or a directory containing image.png, positions.image.csv with headers barcode, x, and y, and optionally diameter.image.txt containing spot diameter
- --markers
Path(s) to JSON file that maps name to features. For example {“a”:[“gene1”, “gene2”], “b”:[“gene3”]}
- --host
Host IP address
- --port
Server port
Default: 5000
- --no-open
Do not open your web browser
Default: False
- --results
URL to save user computed results (e.g. differential expression)
- --ontology
Path to ontology in OBO format for annotation
- --tmap
Path(s) to transport maps directory computed with WOT
Serve
The serve command starts the cirrocumulus server for use in a shared server environment which can handle concurrent requests from multiple users. The server can optionally enforce permissions at the dataset level, in order to securely share datasets with collaborators. Additionally, annotations and sets are shared among all users authorized to view a dataset and are stored in a database. The server can be deployed on a cloud VM, an on-premise machine, or on Google App Engine. When deployed in App Engine, the server can be configured to be use Google Cloud Firestore as a database. On AWS, we recommend using DocumentDB. Please note that no datasets are available until you import a dataset into cirrocumulus.
usage: cirro serve [-h] [--db_uri DB_URI] [-w WORKERS] [-t TIMEOUT] [-b BIND]
[--footer FOOTER] [--header HEADER] [--upload UPLOAD]
[--results RESULTS] [--ontology ONTOLOGY]
Named Arguments
- --db_uri
Database connection URI
Default: “mongodb://localhost:27017/cirrocumulus”
- -w, --workers
The number of worker processes
- -t, --timeout
Workers silent for more than this many seconds are killed and restarted
Default: 30
- -b, --bind
Server socket to bind. Server sockets can be any of $(HOST), $(HOST):$(PORT), fd://$(FD), or unix:$(PATH). An IP is a valid $(HOST).
Default: “127.0.0.1:5000”
- --footer
Markdown file to customize the application footer
- --header
Markdown file to customize the application header
- --upload
URL to allow users to upload files
- --results
URL to save user computed results (e.g. differential expression) to
- --ontology
Path to ontology in OBO format for annotation
Prepare Data
The prepare_data command is used to freeze an h5ad, loom, or Seurat file in cirrocumulus format. The cirrocumulus format allows efficient partial dataset retrieval over a network (e.g. Google or S3 bucket) using limited memory. Please note that when converting Seurat files, the data slot from the default assay is used.
usage: cirro prepare_data [-h] [--out OUT] [--format {parquet,jsonl,zarr}]
[--whitelist WHITELIST] [--markers MARKERS]
[--no-auto-groups] [--groups GROUPS]
[--group_nfeatures GROUP_NFEATURES]
[--spatial SPATIAL]
dataset [dataset ...]
Positional Arguments
- dataset
Path to a h5ad, loom, or Seurat (rds) file
Named Arguments
- --out
Path to output directory
- --format
Possible choices: parquet, jsonl, zarr
Output format
Default: “zarr”
- --whitelist
Optional whitelist of fields to save when output format is parquet or zarr. Use obs, obsm, or X to save all entries for these fields. Use field.name to save a specific entry (e.g. obs.leiden)
- --markers
Path to JSON file of precomputed markers that maps name to features. For example {“a”:[“gene1”, “gene2”], “b”:[“gene3”]
- --no-auto-groups
Disable automatic cluster field detection to compute differential expression results for
Default: False
- --groups
List of groups to compute markers for (e.g. louvain). Markers created with cumulus/scanpy are automatically included. Separate multiple groups with a comma to combine groups using “AND” logic (e.g. louvain,day)
- --group_nfeatures
Number of marker genes/features to include
Default: 10
- --spatial
Directory containing 10x visium spatial data (tissue_hires_image.png, scalefactors_json.json, and tissue_positions_list.csv) or a directory containing image.png, positions.image.csv with headers barcode, x, and y, and optionally diameter.image.txt containing spot diameter
Concatenate
The concat command is used to concatenate multiple datasets into a single dataset. Both spatial and non-spatial embeddings are concatenated in a tiled layout:
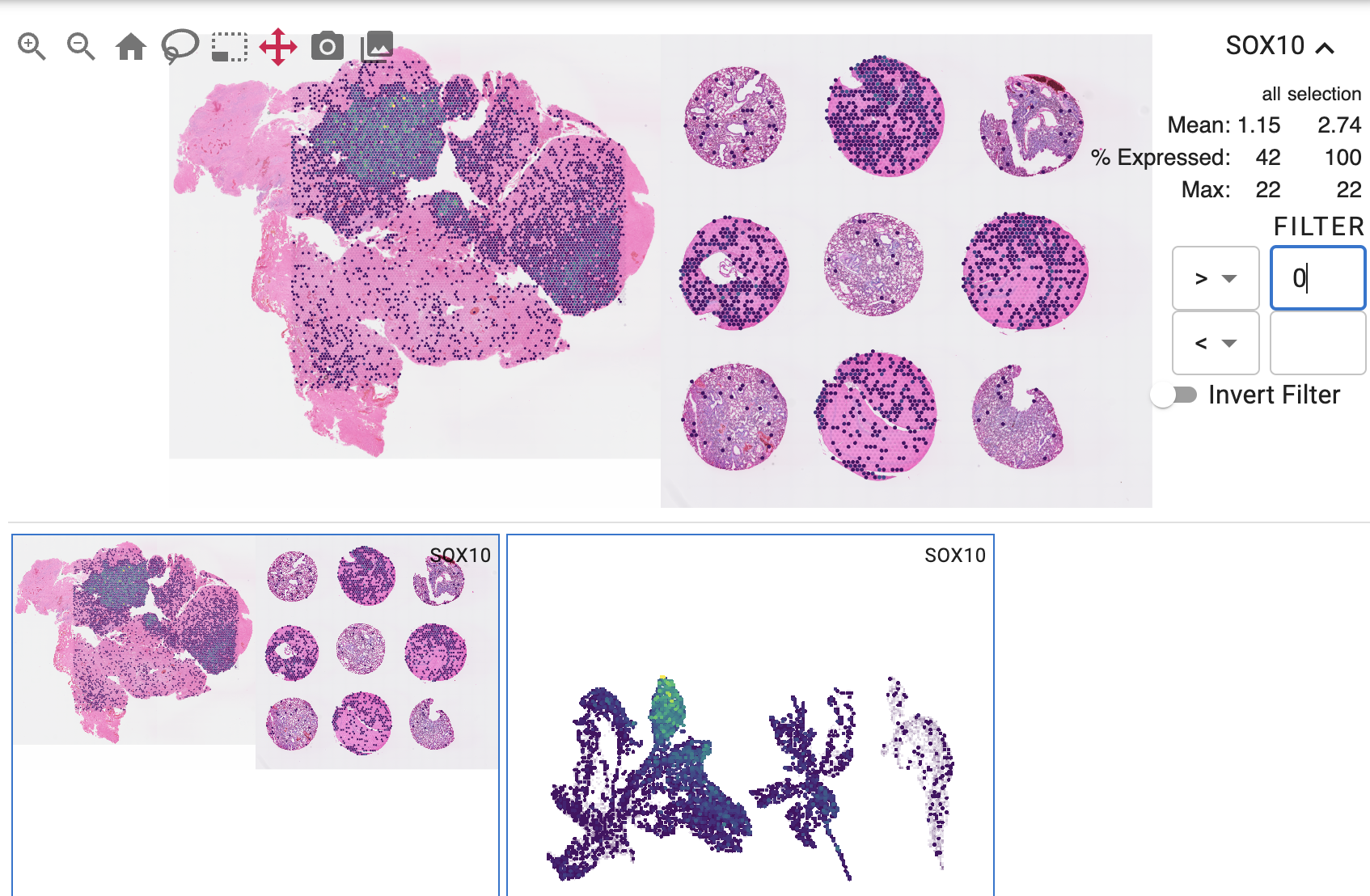
usage: cirro concat [-h] [-o OUTPUT] [-c COLS] dataset [dataset ...]
Positional Arguments
- dataset
Paths to dataset in h5ad, 10x h5, loom, or Seurat (rds) format. For spatial dataset, a ‘spatial’ directory must exist in the same directory as each dataset
Named Arguments
- -o, --output
Output path to write concatenated h5ad file. A spatial directory will be created if all input datasets are spatial
Default: “data-concat.h5ad”
- -c, --cols
Number of columns in tiled layout
Default: 2